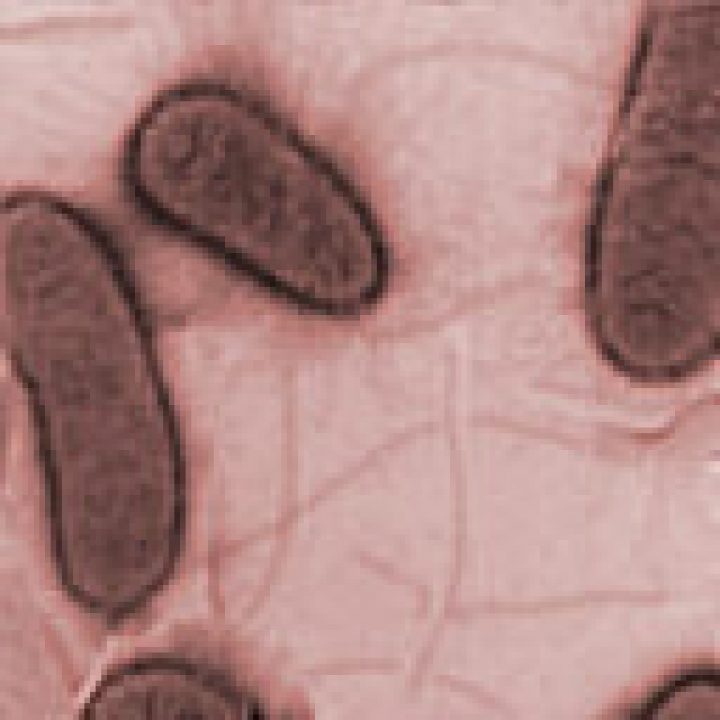
Studie zum EHEC-Ausbruchsstamm: Anheftungsmuster, die einer Mauer aus Backsteinen gleicht
Bakterien, Darminfektion, Durchfall, Durchfall-Erreger, Durchfallerkrankung, EHEC, EHEC-Erreger, Enterohämorrhagische Escherichia coli, Hämolytisch-Urämisches Syndrom, HUS, Infektion
Zirka 4 Wochen nach Eingang der ersten Patientenproben am 23. Mai in Münster erschien am 23. Juni die erste Publikation des Wissenschaftlerteams der Medizinischen Fakultät der Westfälischen Wilhelms-Universität Münster um Prof. Dr. Dr. h.c. Helge Karch. Es handelt sich dabei um eine Arbeit des Instituts für Hygiene und des Instituts für Medizinische Mikrobiologie an der Universität Münster sowie des Nationalen Referenzzentrums für Salmonellen und andere enteritische Pathogene am Robert Koch-Institut (RKI, Aussenstelle Wernigerode). In der Online-Ausgabe des Lancet Infectious Diseases berichten die EHEC-Forscher über ihre Forschungsergebnisse zur geno- und phänotypischen Charakterisierung von insgesamt 80 Patientenisolaten, die aus 17 Städten Deutschlands stammen und in dem Zeitraum vom 23. Mai bis 2. Juni 2011 isoliert wurden.
Zunächst wurde die genetische Verwandtschaft zu dem HUSEC041-Referenzstamm aus dem Jahre 2001 ermittelt. Anschliessend erfolgte die Bestimmung der potentiellen Virulenzfaktoren. Weiterhin wurde die zellschädigende Wirkung aller 80 isolierten Erreger auf Nierenzellen und deren Interaktion mit Darmzellen analysiert. Prof. Karch: Die Ergebnisse zeigen, dass alle Isolate demselben Sequenztyp (ST) 678 zugeordnet werden können. Innerhalb der HUSEC-Kollektion und bei anderen EHEC ist dieser Sequenztyp bislang einmalig gewesen. Er wurde von uns erstmalig 2008 für den Referenzstamm des HUSEC041-Klons veröffentlicht. Danach ist er nicht mehr in Erscheinung getreten. Völlig unerwartet taucht er jetzt in unserer Studie 80 mal in Folge bei Patientenisolaten auf.
Alle Ausbruchsstämme besitzen ein Repertoire von 13 Genen, die für unterschiedliche potentielle Virulenzfaktoren kodieren. Neben dem für EHEC typischen Shiga Toxin 2-kodierenden Gen wurden die Gene für Determinanten nachgewiesen, die für die wässrig-blutigen Durchfälle und den damit einhergehenden Elektrolytverlust verantwortlich sind. Dazu gehören beispielsweise ein für Shigella-Bakterien (altbekannte Durchfallerreger) typisches Enterotoxin und ein schleimauflösendes Enzym. Ausserdem wurden die Gene der für die Anheftung verantwortlichen Fimbrien und Adhäsionsproteine ermittelt.
Im Vergleich zu dem zehn Jahre alten HUSEC041-Isolat hat der Ausbruchsstamm das Gen für das enteroaggregative hitzestabile Enterotoxin (EAST1) verloren. Stattdessen hat er das weit verbreitete Gen für das Fimbrienantigen AAF/I aufgenommen und das in HUSEC041 vorkommende Gen für AAF/III verloren. Neben zusätzlichen Antibiotikaresistenzgenen sind dies die einzigen Unterschiede, die wir bisher gefunden haben. Da all die genannten Gene auf mobilen genetischen Elementen (Plasmiden) liegen, sind solche Aufnahmen und Verluste eher die Regel als die Ausnahme bei pathogenen E. coli , erklärt der Direktor des Instituts für Hygiene am Universitätsklinikum Münster.
Bei ihrer Interaktion mit Darmzellen zeigen alle Stämme das für enteroaggregative E. coli (EAEC) typische Anheftungsmuster, das einer Mauer aus Backsteinen gleicht, sagt Prof. Karch. Aus dem Darm wird das Shiga Toxin 2 über einen noch nicht genau aufgeklärten Mechanismus in den Blutkreislauf transferiert. Dort bindet es bevorzugt an Nieren- und Gehirnendothelzellen. Ersteres verursacht Niereninsuffizienzen und zweiteres kann zu zerebralen Schädigungen führen, was die schweren, unter Umständen tödlichen Krankheitsverläufe erklärt, so Prof. Karch.
Diese Studie sei ein wichtiger Schritt zum Verständnis des Erregers, so Karch. Nun gelte es aber, noch viele weitere Fragen zu beantworten, um das aggressive Verhalten des aktuellen Ausbruchsstamms zu erklären. Karch: Ist es nur die Kombination der krankheitsauslösenden Faktoren, die ihn so aggressiv macht? Ungeklärt bleibt aktuell auch noch die Frage nach der Infektionsdosis, d.h. wie viele Erreger waren auf oder in den Sprossen? Und ist dieser Erreger, der nahezu aus dem Nichts kam, auf eine Humanpopulation gestossen, die keinen Immunschutz aufweist, weil es zuvor noch keinen Kontakt des Menschen mit dem gleichen Erreger gab? In welchem Biotop hat er überlebt, wenn er nicht im Menschen ist? Diese Fragen gilt es zukünftig zu beantworten, um Strategien zu entwickeln und Wege zu finden, wie wir ihn langfristig und nachhaltig nicht nur bekämpfen, sondern letztendlich vollständig eliminieren können.
Quelle: UKM

